
Introduction
Mutagenic impurities, formerly known as genotoxic impurities (GI) or potential genotoxic impurities (PGI), are compounds that can modify DNA and, consequently, can cause cancer. Impurities present in the marketed drug must be evaluated early in the drug development process. To that end, analytical techniques must be developed that are sharp and specific sufficiently to select the levels in both drug import and product.
The International Conference on Harmonization published ICH M7 guidelines, which highlight the requirements for assessing and controlling DNA-reactive impurities to ensure the safety of pharmaceutical products. The European Medicine Evaluation Agency (EMEA), U.S. FDA, and the Asia regulatory agencies follow these guidelines. They require that any mutagenic impurities in a drug substance or product must be below the Threshold of Toxicological Concern (TTC) of 1.5 µg per day based on the Maximum everyday dosage of the pharmaceutical combination over a lifetime. For example, for a dosage of 1 g of Active Pharmaceutical Ingredient (API) per daytime, any impurity must be smaller than 1.5 ppm (1.5 µg). This is orders of magnitude lower than general pharmaceutical impurities analysis, which is at the 500-ppm level and governed by Q3B(R).
What is Methyl 3-Aminocrotonate?
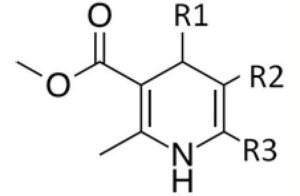
Methyl-3-aminocrotonate (MAC) is a Michael-reactive receptor and a starting material in different cardiovascular drug products. The API used is an active sense from a proprietary pharmaceutical product; therefore, just the partial structure is shown in Figure 1. MAC flags up a favorable from the mutagenic structural attention. This compound would typically be analyzed by static head space (SHS) GC-MS after derivatization with trifluoroacetic anhydride to increase the volatility (Figure 2).3 When MAC (underivatized) was analyzed by UPLC-MS, nothing was noticed; this was supposed to be due to its insufficient stability in aqueous solvents.
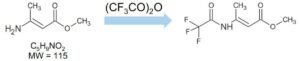
In this type of trace analysis, where there is a large amount of matrix, it would be advantageous if chemical derivatization of the mutagenic impurity could be avoided for the following reasons:
The shape of acylation derivatives can be challenging to prepare
Response bi-products can happen, which could add more complication to the matrix
The different derivatization step would need extra validation to be completed
Benidipine
Benidipine is a manufactured dihydropyridine calcium channel blocker used to dine hypertension and angina pectoris.
Indication
Benidipine is a powerful and long-lasting medicine for treating cardiovascular conditions such as hypertension, renoparenchymal hypertension and angina pectoris.
Pharmacodynamics
Benidipine reduces systolic and diastolic blood pressure and presents a decrease in heart rate pulse after treatment. It is also reported a decrease in urinary protein excretion and serum triglycerides. Different studies have shown benidipine anti-oxidative action, stimulation of NO production, suppression of bonding molecules presentation, stimulation of osteoblast differentiation, and suppression of the increase of vascular soft muscle cells and mesangial cells as myocardial saving. The enhancement of NO production is associated with benidipine’s cardioprotective and antiatherosclerotic effects.
Absorption
Benidipine is rapidly spread in oral administration, reaching the highest concentration within 2 hours. The short time needed for maximum concentration to reach a particular characteristic of benidipine compared with other calcium channel blockers. The registered maximum concentration and AUC are dose-dependent, and they can go from 0.55-3.89 ng/ml and 1.04-6.7 ng.h/ml, respectively, when administered at a dose of 2-8 mg.
Volume of distribution
Benidipine is highly distributed to the tissues mostly in the liver, kidneys, and plasma. It does not offer a high expansion following repeated oral administrations.
Side Effects of BENIDIPINE
- Fatigue
- Ankle swelling
- Sleepiness
- Flushing (sense of warmness in the front, ears, neck and chest)
- Headache
- Dizziness
- Palpitations
- Nausea
- Oedema (swelling)
- Abdominal pain
Cilnidipine
Cilnidipine is a dihydropyridine calcium channel blocker with activity on both N- and L-type calcium media used to treat hypertension.
Indication
Cilnidipine is suggested for the management of hypertension for end-organ security. It is reported to be useful in elderly patients and those with diabetes and albuminuria. Cilnidipine has been increasingly utilized in patients with chronic kidney disease. Hypertension is the phrase used to describe the existence of high blood pressure. Blood stress is generated by the power of the blood pumped from the heart against the blood vessels. Thus, hypertension is generated when too much pressure is on the blood vessels, damaging the blood vessel.
Pharmacodynamics
The administration of cilnidipine has been shown to present an antisympathetic profile in vitro and in vivo. It decreases blood pressure carefully and effectively without excessive blood pressure decrease or tachycardia.
Absorption
Cilnidipine offers an extremely rapid absorption with a highest peaked concentration after 2 hours. Its distribution tends to be higher in the liver and kidneys, plasma and other tissues. Cilnidipine does not offer a high accumulation in the tissue following repeated oral administration. Cilnidipine is reported to offer very low bioavailability, approximately 13%. This low bioavailability is suggested due to its low aqueous solubility and high permeability. Hence, efforts have been made to find an innovative formulation that can significantly improve the bioavailability of this drug. One of these formulations compares the generation of polymeric nanoparticles, which improves the bioavailability by 2.5-3-fold.
Volume of distribution
Drugs in the group of dihydropyridines, such as cilnidipine, tend to have a large volume of distribution.
Side Effects of CILNIDIPINE
- Headache
- Feeling exhausted
- Swollen ankles
- Nausea
Isradipine
Isradipine is a dihydropyridine calcium drain blocker utilized for the therapy of hypertension.
Pharmacodynamics
Isradipine decreases arterial soft muscle contractility and subsequent vasoconstriction by impeding the influx of calcium ions through L-type calcium media. Calcium ions joining the cell through these channels bind to calmodulin. Calcium-bound calmodulin then attaches to and activates myosin light chain kinase (MLCK). Activated MLCK catalyzes the phosphorylation of myosin’s regulatory light chain subunit, a key phase in muscle contraction. Signal amplification is performed by calcium-induced calcium departure from the sarcoplasmic reticulum through ryanodine receptors. Inhibition of the initial inflow of calcium decreases the contractile action of arterial soft muscle cells and results in vasodilation. The vasodilatory results of isradipine result in a general decrease in blood pressure.
Absorption
Isradipine is 90%-95% absorbed and is subject to extensive first-pass metabolism, resulting in a bioavailability of about 15%-24%.
Toxicity
Symptoms of overdose include lethargy, sinus tachycardia, and temporary hypotension. Significant lethality was followed in mice presented with oral doses of around 200 mg/kg, and rabbits provided about 50 mg/kg of isradipine. Rats tolerated amounts of over 2000 mg/kg without consequence in survival.